
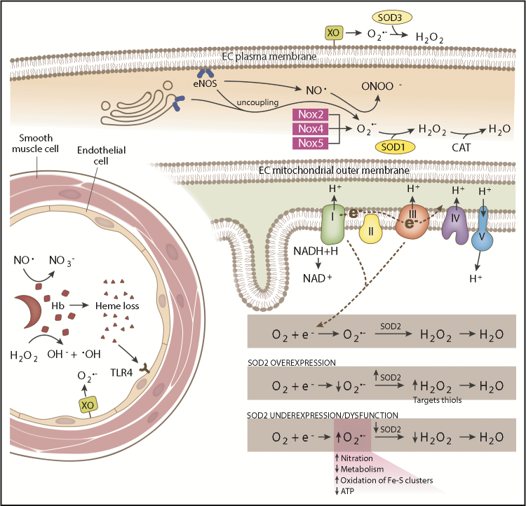
 From the article in Blood Advances: Sickle cell disease (SCD) is an inherited hemoglobinopathy caused by a single point mutation in the β-globin gene. As a consequence, deoxygenated hemoglobin polymerizes triggering red blood cell sickling and hemolysis, vaso-occlusion, and ischemia/reperfusion. Allied to these pathologies is the overproduction of reactive oxygen species driven by hemoglobin Fenton chemistry and peroxidase reactions as well as by secondary activation of vascular oxidases, including NAD(P)H oxidase and xanthine oxidase. In addition, hypoxia, produced by sickle red blood cell occlusion, disrupts mitochondrial metabolism and generates excess superoxide through electron leak from the mitochondrial respiratory chain. Superoxide dismutase 2 (SOD2) is a mitochondrial-specific antioxidant enzyme that dismutates superoxide to hydrogen peroxide, which is then converted to water by catalase and glutathione peroxidase. In SCD, the antioxidant defense system is significantly diminished through decreased expression and activity levels of antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase. From a translational perspective, genetic variants including a missense variant in SOD2 (valine to alanine at position 16) are present in 45% of people with African ancestry and are associated with increased sickle complications. While it is known that there is an imbalance between oxidative species and antioxidant defenses in SCD, much more investigation is warranted. This review summarizes our current understanding of antioxidant defense systems in SCD, particularly focused on SOD2, and provides insight into challenges and opportunities as the field moves forward.
From the article in Blood Advances: Sickle cell disease (SCD) is an inherited hemoglobinopathy caused by a single point mutation in the β-globin gene. As a consequence, deoxygenated hemoglobin polymerizes triggering red blood cell sickling and hemolysis, vaso-occlusion, and ischemia/reperfusion. Allied to these pathologies is the overproduction of reactive oxygen species driven by hemoglobin Fenton chemistry and peroxidase reactions as well as by secondary activation of vascular oxidases, including NAD(P)H oxidase and xanthine oxidase. In addition, hypoxia, produced by sickle red blood cell occlusion, disrupts mitochondrial metabolism and generates excess superoxide through electron leak from the mitochondrial respiratory chain. Superoxide dismutase 2 (SOD2) is a mitochondrial-specific antioxidant enzyme that dismutates superoxide to hydrogen peroxide, which is then converted to water by catalase and glutathione peroxidase. In SCD, the antioxidant defense system is significantly diminished through decreased expression and activity levels of antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase. From a translational perspective, genetic variants including a missense variant in SOD2 (valine to alanine at position 16) are present in 45% of people with African ancestry and are associated with increased sickle complications. While it is known that there is an imbalance between oxidative species and antioxidant defenses in SCD, much more investigation is warranted. This review summarizes our current understanding of antioxidant defense systems in SCD, particularly focused on SOD2, and provides insight into challenges and opportunities as the field moves forward.
https://ashpublications.org/bloodadvances/article/3/17/2679/261359/Decoding-the-...